
Bioäquivalenz – Das zentrale Konzept der Arzneimittelentwicklung
Im Rahmen der Zulassung eines generischen Arzneimittels, muss dessen Bioäquivalenz zum Originatorprodukt belegt werden – soweit, so bekannt. Aber wussten Sie, dass auch Originatoren Bioäquivalenzstudien durchführen?

Bedeutung, Folgen und Sinn des Bioäquivalenzkonzepts
Der Grund ist in beiden Fällen der gleiche: Bioäquivalenz liefert den Nachweis, dass 2 Präparate ein vergleichbares pharmakokinetisches in-vivo-Verhalten zeigen. Damit darf im Rahmen eines Zulassungsverfahrens, auf die Daten zur Sicherheit und Wirksamkeit des Referenzpräparates Bezug genommen werden.
Im Fall von generischen Arzneimitteln ist eine Bioäquivalenzprüfung der Nachweis, dass ein Originatorprodukt gefahrlos gegen ein Generikum ausgetauscht werden kann. Der Gesetzgeber geht an dieser Stelle sogar noch weiter und fordert, dass sich Generikum und Originatorprodukt in praktisch allen Belangen vergleichbar verhalten.
Beispielsweise ist vorgeschrieben, dass die Stärke beider Arzneimittel identisch sein muss, obwohl es bei manchen Arzneistoffen möglich wäre, trotz niedrigerer Stärke die gleiche Bioverfügbarkeit wie der Originator zu erreichen. Damit könnte z. B. die Menge unresorbierten Arzneistoffs, der mit dem Fäzes in die Umwelt gelangt, reduziert werden.
Ein weiteres Beispiel: Zeigt ein Originatorprodukt unterschiedliche Bioverfügbarkeiten in Abhängigkeit von den Mahlzeiten, dann muss auch ein Generikum ein statistisch gesehen äquivalentes Verhalten zeigen. Dabei würde eine von den Mahlzeiten unabhängige Bioverfügbarkeit die korrekte Medikamenteneinnahme erleichtern.
Der Sinn hinter dieser Regelung ist, dass Arzt, Apotheker und Patient beim Wechsel zwischen Originatorprodukt und Generikum nicht umdenken müssen und damit Verunsicherungen und insbesondere Verwechselungen ausgeschlossen werden. Gleichzeitig ist die Vorsichtsmaßnahme aber auch der Grund, warum es keine Generika mit verbesserten Bioverfügbarkeitseigenschaften gibt.
Auch Originatoren nutzen das Bioäquivalenzkonzept
Aber nicht nur für Generikahersteller ist das Bioäquivalenzkonzept essentiell, auch Originatoren führen Bioäquivalenzprüfungen durch. Originatoren müssen ihre Produkte schnell zur Marktreife bringen, um möglichst hohe Gewinne innerhalb des Patentschutzzeitraumes zu erzielen. Deshalb werden zu Beginn der Entwicklung oft Studien mit nicht finalen Formulierungen durchgeführt. Beispielweise werden erste Studien häufig mit Kapselformulierungen durchgeführt, das endgültige Arzneimittel ist aber oft eine Tablette. Damit der Originator diese früheren Daten auch für geänderte Formulierungen nutzen darf, muss er die Bioäquivalenz der Präparate nachweisen.
Bioäquivalenz spielt damit für alle Pharmafirmen eine zentrale Rolle – ganz gleich, ob Originator oder Generikahersteller. Aber was genau bedeutet eigentlich Bioäquivalenz und wie misst man diese?
Alle praxisrelevanten Informationen zur Bioäquivalenz
Wir haben uns die „GUIDELINE ON THE INVESTIGATION OF BIOEQUIVALENCE“ der Europäischen Arzneimittelagentur angeschaut und erläutern Ihnen am einfachsten Fall, einer schnellfreisetzenden Tablette, die wichtigsten Fakten.
Die komplette Guideline steht für interessierte Leser auf der Homepage der EMA als Download bereit.
Definition der Bioäquivalenz - Was ist Bioäquivalenz?
Entsprechend der Definition der Europäische Arzneimittelagentur sind 2 Arzneimittel dann bioäquivalent, wenn deren Bioverfügbarkeiten hinsichtlich Geschwindigkeit und Ausmaß nach Administration der gleichen molaren Dosis innerhalb akzeptierter vordefinierter Grenzen liegen.
Geschwindigkeit und Ausmaß mit der der Arzneistoff ins Blut gelangt, werden an Hand von Plasmakonzentrations-Zeit-Diagrammen (Abbildung 1) bestimmt. Dazu werden die im Blutplasma gemessenen Wirkstoffkonzentrationen gegen die jeweiligen Blutentnahmezeitpunkte aufgetragen.

Abbildung 1: Idealisiertes Konzentrations-Zeit-Diagramm.
Ein solches Konzentrations-Zeit-Diagramm weist 3 charakteristische Werte auf.
- Die Fläche unter Kurve (AUC) steht für das Ausmaß der Wirkstoffaufnahme.
- Die maximale Konzentration (Cmax) sowie die Zeit bis zum Erreichen dieser maximalen Konzentration (tmax) stehen für die Geschwindigkeit der Wirkstoffaufnahme. In der Regel wird jedoch nur Cmax betrachtet.
Für die Beurteilung der Bioäquivalenz werden die Quotienten von AUC und Cmax des Test- und Referenzpräparates verglichen (Abbildung 2). Beträgt dieser Wert 100 % sind die Werte beider Präparate exakt gleich, bei Werten < 100 % ist der Wert des Testpräparates kleiner und bei Werten > 100 % größer als der des Referenzpräparates. Allerdings erfordert die Variabilität der Messwerte eine etwas komplexere statistische Auswertung, wie später noch gezeigt wird.
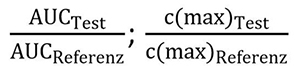
Abbildung 2: Berechnung der Bioäquivalenz-Quotienten.
Die Zeit bis zum Erreichen der maximalen Plasmakonzentration wird nur dann verglichen, wenn ein schnelles Anfluten klinisch relevant für Wirkungen oder Nebenwirkungen ist.
Der Mensch ist keine Maschine – Von den Schwierigkeiten der Bioäquivalenzbestimmung
Das Konzept der Bioäquivalenz ist simpel und nachvollziehbar. Allerdings kann die Bestimmung der Bioäquivalenz einige Schwierigkeiten mit sich bringen.
In der pharmazeutischen Industrie ist es Standard, ausschließlich Analytik-Methoden zu verwenden, die nachweislich zu reproduzierbaren Ergebnissen mit sehr kleinen Messungenauigkeiten führen.
Für die Bestimmung der Bioäquivalenz stehen solche Methoden jedoch nicht zur Verfügung. Denn trotz aller Bemühungen gibt es bis heute kein in-vitro-Testverfahren, dass zuverlässig den menschlichen Organismus simulieren kann. Deshalb wird die Bioäquivalenz mit Hilfe freiwilliger Testpersonen bestimmt. Allerdings zeigen biologische Systeme immer Variabilität und folglich zeigt auch die Bioverfügbarkeit von Arzneimitteln intra- und interindividuelle Unterschiede – suboptimale Bedingungen, wenn es darum geht, Unterschiede aufzudecken, die allein auf das Arzneimittel zurückzuführen sind.
Um die Bestimmung der Bioäquivalenz dennoch zu ermöglichen, werden statistische Testverfahren sowie ein standardisiertes Testprotokoll eingesetzt.
Bioäquivalenzstudien
Standardisierung der Testpersonen
Ein wichtiger Schritt zur Standardisierung und gleichzeitigen Reduzierung der Variabilität der Bioverfügbarkeit ist die Rekrutierung der Testpersonen.
In der Regel werden gesunde Probanden rekrutiert, sofern dies auf Grund von Sicherheitsbedenken nicht ausgeschlossen wird. Birgt die Einnahme eines Arzneimittels hohe gesundheitliche Risiken werden Patienten für die Studien rekrutiert.
Um geeignete Probanden zu rekrutieren, werden alle Kandidaten medizinisch untersucht und ihre medizinische Historie geprüft. Die Auswahl erfolgt dann auf Grundlage definierter Ein- und Ausschlusskriterien.
Meist sind die Probanden männlich, haben einen Body Mass Index zwischen 18,5 – 30 kg/m², nehmen keine weiteren Arzneimittel ein, sind Nichtraucher, nehmen keine Drogen und hatten in der Vergangenheit keine Alkoholprobleme. Auch Genotypisierungen sind möglich, um z. B. Langsam- und Schnell-Metabolisierer auszuschließen.
Standardisierung des Studienprotokolls
Die Guideline der EMA empfiehlt weiterhin Parameter zu standardisieren, die Einfluss auf die Wirkstoffaufnahme haben können. Dazu gehören die Nahrungsmittelaufnahme, die Flüssigkeitszufuhr, die körperliche Aktivität sowie die Körperhaltung, also Stehen, Sitzen oder Liegen.
Die Durchführung der Bioverfügbarkeitsstudien erfolgt in der Regel nüchtern. Sofern das Originator-Arzneimittel jedoch ausdrücklich zum Essen eingenommen werden soll, erfolgt die Einnahme zusammen mit einer ebenfalls standardisierten Mahlzeit.
Die Planung, Durchführung und Auswertung
Aller Standardisierungsbemühungen zum Trotz, ist bei Bioäquivalenzstudien eine recht große Variabilität zu beobachten. Um dennoch valide Schlussfolgerungen zur Bioäquivalenz machen zu können, werden Bioäquivalenzstudien in allen Phasen, von der Planung bis zur Auswertung statistisch gestützt.
Planung
In der Planungsphase berechnen Statistiker die notwendige Anzahl an Probanden, um eine ausreichende statistische Power der Studie zu gewährleisten. In diese Berechnung fließt z. B. die Variabilität der Pharmakokinetik des Wirkstoffs ein. Diese kann je nach Wirkstoff unterschiedlich sein und ist z. B. aus der Literatur bekannt. Die Probandenanzahl sollte in jedem Fall größer als 12 sein.
Durchführung
Die Durchführung von Bioäquivalenzstudien erfolgt in der Regel als randomisierte Crossover-Studie, um intra-individuelle Einflussfaktoren zu minimieren. Das bedeutet, dass jeder Proband im Verlauf der Studie sowohl das Test- als auch das Referenz-Arzneimittel einnimmt (Abbildung 3). Dazu wird zufällig zugeteilt, welches Arzneimittel die Probanden in der ersten Testphase einnehmen. In der zweiten Testphase nehmen sie dann das andere Arzneimittel ein. Zwischen beiden Testphasen liegt eine sogenannte Auswaschphase, in der der Wirkstoff weitgehend aus dem Körper eliminiert wird. Sie beträgt mindestens 5 Halbwertszeiten.
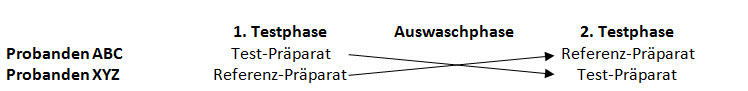
Abbildung 3: Schema des Crossover-Studiendesigns.
Auswertung
Für die Ergebnis-Auswertung der Bioäquivalenz wird die ANOVA-Datenanalyse empfohlen, um das 90 %-Konfidenzintervall der Quotienten der AUC- und cmax-Werte jeweils von Test- und Referenzpräparat zu bestimmen. Das 90 %-Konfidenzintervall besagt, dass der tatsächliche (aber unbekannte) Wert mit einer Wahrscheinlichkeit von 90 % innerhalb des berechneten Intervalls liegt.
Bioäquivalenz ist nach Guideline dann gegeben, wenn das 90 %-Konfidenzintervall zwischen 80 % und 125 % liegt. Dieses Ziel muss sowohl für die AUC als auch für cmax erreicht werden. Die asymmetrischen Grenzen sind darin begründet, dass es sich um nicht normalverteilte Daten handelt, die während der Auswertung transformiert und zum Schluss wieder zurücktransformiert werden müssen.

Abbildung 4: |--|= 90 %-Konfidenzintervall. Einige fiktive Beispiele veranschaulichen die Kriterien für Bioäquivalenz: Das berechnete 90 %-Konfidenzintervall des Prüfpräparates muss innerhalb der geforderten Grenzen von 80 % - 125 % liegen.
Es gibt auch Abweichungen von diesen Standardgrenzen. Bei Arzneimitteln mit geringer therapeutischer Breite werden die Grenzen auf 90,00 % – 111,11 % eingeengt. Bei Arzneistoffen mit einer sehr hohen intra-individuellen Variabilität können die Grenzen auch erweitert werden.
Fazit
Sorgfältige Planung, hochentwickelte Analytik sowie statistische Auswerteverfahren ermöglichen die Überprüfung der Bioäquivalenz von Arzneimitteln. Zwei bioäquivalente Präparate können gegeneinander ausgetauscht werden ohne die Wirksamkeit oder Sicherheit einer Therapie zu gefährden. Bioäquivalenz ist damit eines der zentralen Konzepte in der Arzneimittelentwicklung.

Dr. rer. nat. Michael Rottke beschäftigt sich als promovierter Apotheker besonders gern mit medizinischen und pharmazeutischen Themen. Vor seinem Einstieg ins Marketing arbeitete er mehrere Jahre als Scientist und Laborleiter in der Arzneimittelforschung und -entwicklung der ratiopharm GmbH.
Sie möchten Zugriff auf den gesamten Fachkreis?
Loggen Sie sich ein und erhalten Zugang zu allen Artikeln des Fachkreises
Die folgenden Inhalte richten sich an Mitglieder medizinischer Fachkreise, bitte loggen Sie sich mit Ihrem kostenlosen ratiopharm Benutzerkonto oder Ihrem DocCheck Account ein.
Sie sind nicht im Gesundheitswesen tätig? Besuchen Sie die ratiopharm.de und entdecken Sie spannende Inhalte!